Super-Resolution Microscopy
Recommended Reading:
- https://www.microscopyu.com/techniques/super-resolution
- Super-resolution microscopy at a glance.pdf
- Super-resolution microscopy demystified.pdf
- https://bitesizebio.com/20978/the-whos-who-of-super-resolution-microscopy-part-1/
Background
The excerpt below is taken from: https://en.wikipedia.org/wiki/Super-resolution_microscopy
Super-resolution microscopy, in light microscopy, is a term that gathers several techniques, which allow images to be taken with a higher resolution than the one imposed by the diffraction limit.[1][2] Due to the diffraction of light, the resolution in conventional light microscopy is limited, as stated (for the special case of widefield illumination) by Ernst Abbe in 1873.[3] In this context, a diffraction-limited microscope with numerical aperture N.A. and light with wavelength λ reaches a lateral resolution of d = λ/(2 N.A.) - a similar formalism can be followed for the axial resolution (along the optical axis, z-resolution, depth resolution). The resolution for a standard optical microscope in the visible light spectrum is about 200 nm laterally and 600 nm axially.[4] Experimentally, the attained resolution can be measured from the full width at half maximum(FWHM) of the point spread function (PSF) using images of point-like objects. Although the resolving power of a microscope is not well defined,[5] it is generally considered that a super-resolution microscopy technique offers a resolution better than the one stipulated by Abbe.
Super-resolution imaging techniques rely on the near-field (photon tunneling microscopy[6] as well as those that utilize the Pendry Superlens and near field scanning optical microscopy) or on the far-field. Among the latter are techniques that improve the resolution only modestly (up to about a factor of two) beyond the diffraction-limit like the confocal microscope (with closed pinhole), or confocal microscopy aided with computational methods such as deconvolution[7] or detector-based pixel reassignment (e.g. AiryScan, re-scan microscopy,[8] pixel reassignment [9]), the 4Pi microscope, and also structured illumination microscopy technologies like SIM and SMI.
There are two major groups of methods for super-resolution microscopy in the far-field that can improve the resolution with a much larger factor:[10]
- Deterministic super-resolution: The most commonly used emitters in biological microscopy, fluorophores, show a nonlinear response to excitation, and this nonlinear response can be exploited to enhance resolution. These methods include STED, GSD, RESOLFT and SSIM.
- Stochastic super-resolution: The chemical complexity of many molecular light sources gives them a complex temporal behavior, which can be used to make several close-by fluorophores emit light at separate times and thereby become resolvable in time. These methods include Super-resolution optical fluctuation imaging (SOFI) and all single-molecule localization methods (SMLM) such as SPDM, SPDMphymod, PALM, FPALM, STORM and dSTORM.
On October 8, 2014, the Nobel Prize in Chemistry was awarded to Eric Betzig, W.E. Moerner and Stefan Hell for "the development of super-resolved fluorescence microscopy," which brings "optical microscopy into the nanodimension".[11][12]
Stimulated emission depletion (STED) Microscopy
IMB Systems
Excerpt below taken from: https://bitesizebio.com/21988/the-power-of-sted-microscopy-part-1-how-does-it-work/
How STED works… in detail!
A STED microscope is built on the basis of a confocal laser-scanning microscope. For those that do not know everything about a confocal laser-scanning microscope off the top of their head here is a brief reminder:
Confocal laser-scanning microscopy works when an objective focuses a laser light onto a small spot on your sample causing all fluorophores within said spot to emit fluorescence. The light intensity of which distributes according to the point spread function (PSF) and limits the resolution of the image. This emitted fluorescence is then collected by the objective and sent to the detector, which is outputted as a single pixel. To collect more pixels the position of this focused spot needs to be moved. By either moving the ‘scanning mirror’ within the microscope, or the sample itself. This ‘scanning’ is what allows an image to be built up.
In contrast to confocal laser-scanning microscope, in STED microscopy a second laser has been added. Now during image acquisition, the normal excitation laser pulse is closely followed by a doughnut-shaped pulse of a longer wavelength, termed the STED beam. The excited fluorophores that are exposed to the STED beam are instantaneously ‘bleached’ back to the ground state. Therefore, only molecules that are sitting in the centre of the STED beam (the hole in the doughnut) are able to emit fluorescence. This physically narrows the PSF, therefore increasing resolution beyond the diffraction limit.

Principles of STED microscopy
AiryScan Microscopy
IMB Systems
Excerpt below taken from: https://bitesizebio.com/product_article/zeiss-airyscan-a-brave-new-microscopy-world-with-sharper-confocal-resolution/
Typically, confocal microscopes allow light only from one point to reach the detector using a special kind of aperture, called a pinhole. Light coming from planes above or below the focal plane is out of focus when it reaches the pinhole. So, most of it cannot pass the pinhole, and does not contribute to the formation of the image.
Too large a pinhole, then too much light or “noise” gets through; too small a pinhole, then not enough light or “signal” gets detected. The pinhole size must maximize the signal- to-noise ratio of the image. By varying the pinhole diameter, the degree of confocality can be adapted to best fit – usually at one Airy Unit (1 AU).
There has always been a trade off in confocal microscopy among three parameters: sampling frequency (speed), sampling frequency and pinhole size (sensitivity) and pinhole size (resolution). This gives us this “triangle” of parameters, which are interconnected in such a way that no net gain can be made without a sacrifice. For example, if a smaller aperture is used and this is compensated by increased excitation, then other issues arise— such as photobleaching or phototoxicity.
Improving on the Airyscan Principle
Using a physical aperture for the pinhole has been a major limitation for the filed of confocal microscopy. However, an acentric and shifted pinhole detector can produce the same image as the normal pinhole, but smaller in amplitude and shifted in space. This led to the introduction of the ZEISS Airyscan detector; an array of detector elements that would use, and not reject, the light outside the area of 1 AU.
ZEISS LSM 880 with Airyscan: Beam Path and Principle of Airyscanning
Incident light is collected by a hexagonal micro lens array that connects directly to a 32 high transmission optical fibers. These fibers then project to a linear GaAsP-PMT array of the Airyscan detector. In this way, the Airy disk is imaged via zoom optics on to the detector array and no pinhole is needed, as the single detector elements act as separate pinholes. The detection efficiency is increased by reassigning the detected photons from the array elements to the central detection position and summing up the signals from all detector elements of the array. Instead of rejecting some of the light, as in conventional pinhole-based systems, all the light is collected and reassigned to the correct region, resulting in increased signal levels.
ZEISS Airyscan is the future of confocal microscopy
The new ZEISS Airyscan detector geometry maximizes the signal-to noise ratio and gives the same resolution benefits as a pinhole setting of 0.2 AU with the light collection efficiency of a 1.25 AU pinhole. The diameter of the Airyscan detector comprises 6 detector elements, each representing a pinhole of the size of the imaged Airy disk divided by 6. Hence, as the zoom optics are set to capture 1.25 AU, each single detector element act as a pinhole with a size of 1.25 AU/6 = 0.2 AU. This allows each image to be individually deconvolved, further enhancing resolution, and photons can be precisely reassigned by software, resulting in a lateral resolution enhancement by a factor of 1.7. Indeed, this results in overall increased resolution: 140 nm lateral and 400 nm axial resolution, at 488 nm. The resulting image signal-to-noise ratio is drastically improved when comparing to traditional confocal imaging with a 0.2 AU pinhole. In fact, it achieves a 4 fold increase over conventional confocal systems. Taken together, the new ZEISS Airyscan detector can allow you to work on previously challenging optically dense materials.
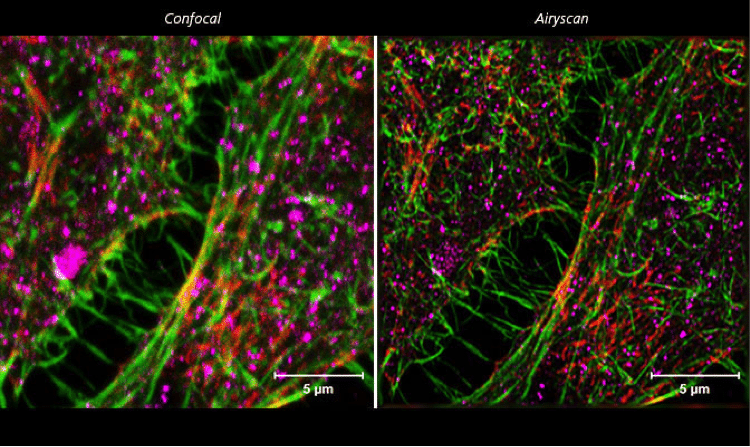
Figure 2. ZEISS LSM 880 with Airyscan: HeLa cells. Conventional confocal microscopy versus Airyscanning of HeLa cells. Sample courtesy of S. Traikov, BIOTEC, TU Dresden, Germany.
For the first time, the ZEISS Airyscan detector has been able to get past microscopy’s three-sided compromises of image resolution, by replacing the most fundamental part of a laser scanning microscope: the pinhole. You can have higher speed, higher resolution, or higher sensitivity—or any combination of the three to the desired degree. With the ZEISS Airyscan detector you can exploit the full potential of your confocal microscope.